
はじめに
こんにちは、富士通研究所 コンピューティング研究所の吉本勇太です。私たちは、高精度分子動力学シミュレーション向けニューラルネットワークポテンシャルの自動生成ツールGeNNIP4MD [1] の開発に取り組んでいます。なお、GeNNIP4MDの詳細に関しては、前回のMaterials Informatics特集 #6で詳しく解説していますので、ご興味がある方は以下からご一読ください。
今回のMaterials Informatics特集 #7では、GeNNIP4MDを用いて作成した高分子電解質膜(ナフィオン)向けニューラルネットワークポテンシャルに関するプレプリント [2] の内容についてご紹介します。
プレプリントの要約
近年、機械学習原子間ポテンシャル(Machine Learning Interatomic Potential, MLIP)は、電子状態計算に匹敵する精度を維持しながら、原子数の多い大規模系での長時間の分子動力学(Molecular Dynamics, MD)シミュレーションを可能にする手法として注目を集めています。しかしながら、MLIPの課題として、従来の古典ポテンシャルに比べて、MDシミュレーション実行時に解析対象の材料構造が崩壊してしまうといった、シミュレーションの安定性の低さが挙げられます。不均質性の高い系やアモルファス材料の解析には、大規模かつ長時間のシミュレーションが必要となる場合が多く、安定したMDシミュレーションを可能にするロバストなMLIPの開発が求められています。本研究では、私たちが開発を進めているMD向けニューラルネットワークポテンシャル生成ツールGeNNIP4MD(Generator of Neural Network Interatomic Potential for Molecular Dynamics)を用いて、広範囲の含水率にわたる高分子電解質膜(ナフィオン)の大規模・長時間MDシミュレーションを可能にするニューラルネットワークポテンシャル(Neural Network Potential, NNP)を構築しました。GeNNIP4MDによりNNPモデルのロバスト性を大幅に向上させることができ、約10,000から20,000原子までの大規模なナフィオン系において、31ナノ秒の長時間の安定したMDシミュレーションが可能になりました。構築したNNPモデルを用いたMDシミュレーションでは、従来の小規模第一原理MDシミュレーションと比較して、広範囲の含水率にわたって、水素原子の自己拡散係数が実験値と良好な一致を示しました。
導入
MLIPを用いたMDシミュレーションは、電子状態計算に匹敵する精度を維持しながら、より大規模な系を原子レベルで解析できる手法として注目を集めています。MLIPは、機械学習モデルを密度汎関数理論(Density Functional Theory, DFT)や波動関数理論などの電子状態計算により生成されたデータセットで訓練することで、従来の古典ポテンシャルよりも多次元ポテンシャルエネルギー表面をより正確に表現できます。一般的に、機械学習モデルの推論にかかる計算時間は、DFTといった電子状態計算による計算時間よりも高速です。そのため、MLIPを活用することで、電子状態計算では計算時間が数年規模になってしまう大規模系の高精度・長時間MDシミュレーションが可能になります。これまでに、カーネル法ベースのポテンシャルやNNP、グラフニューラルネットワークやTransformerに基づくより洗練されたアプローチなど、様々なMLIPが提案されています。これらの適用先は、無機材料、分子系、溶液、高分子材料、界面系など幅広い系に及び、多様な系におけるMLIPの有効性が示されています。
このように、MLIPの材料シミュレーションへの適用は急速に進んでいますが、大規模・長時間MLIP-MDシミュレーションの安定性に焦点を当てた研究は限られています。従来の古典ポテンシャルとは異なり、典型的なMLIPは、物理的な結合/非結合相互作用項を欠いています。その結果、訓練データセットで十分に表現されていない局所原子環境がMLIP-MDシミュレーション中に現れると、原子に作用する力の予測精度が著しく低下し、物理的にあり得ないトラジェクトリが生成されることで、最終的にはシミュレーションの崩壊につながる可能性があります。この安定性の問題は、欠陥を含む系やアモルファス系などの不均質な系では特に重要であり、系の不均質性と遅い動的過程を捉えるためには、大規模・長時間MDシミュレーションが不可欠です。したがって、MLIPモデルの訓練データを生成する際には、多様な構造をサンプリングし、本番のMD実行中に現れる可能性のある局所原子環境を十分に網羅するデータセットを構築することが不可欠です。
私たちは、能動学習(Active Learning, AL)により多様な構造を網羅するデータセットを生成することにより、大規模・長時間MDシミュレーションに適用可能なロバストなNNPを構築できるソフトウェアGeNNIP4MD [1] を開発しています。図1に示すGeNNIP4MDでは、構造サンプリング、スクリーニング、電子状態計算によるラベリング、モデル訓練、精度評価など、NNP構築に関わる複雑な手順を効率的に実行することが可能です。さらに、非平衡NNP-MDシミュレーションと能動学習の組み合わせにより、非平衡構造の効率的なサンプリングを可能にし、長時間のシミュレーションに対するNNPのロバスト性を向上させることができます。
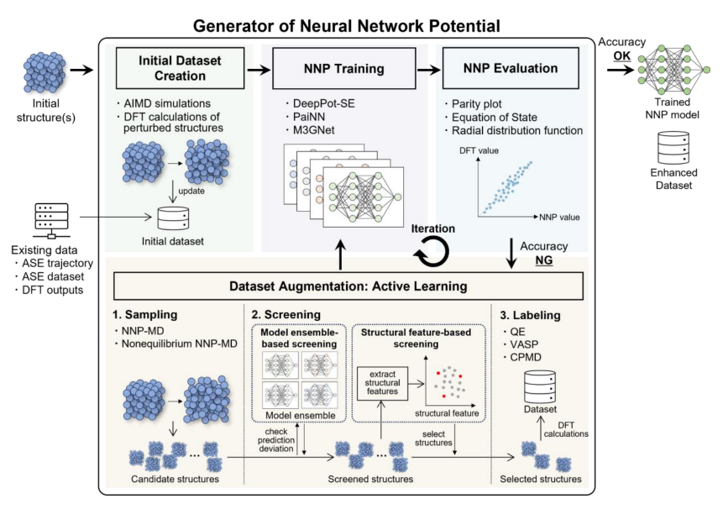
本研究では、GeNNIP4MDを用いて、高分子電解質膜であるナフィオンのNNPモデルを構築し、約10,000から20,000原子までの大規模なナフィオン系において、長時間のNNP-MDシミュレーションを実行しました。ナフィオン膜は、含水率に依存して多様かつ不均質性の高いモルフォロジーを示すため、ナフィオン膜中のプロトン輸送を正確に捉えるためには、大規模・長時間MDシミュレーションが不可欠です。本研究では、GeNNIP4MDにより生成されたNNPモデルを用いて、大規模ナフィオン系のMDシミュレーションを実行し、原子に作用する力の予測値の不確実性に基づいてシミュレーションの安定性を評価しました。不確実性の評価結果に基づいてデータセットを拡張し、NNPを再訓練して、大規模・長時間MDシミュレーションに適したロバストなNNPモデルを構築しました。最終的に得られたNNPモデルを用いて、ナフィオン系の安定した大規模NNP-MDシミュレーションを31ナノ秒の長期間にわたって実行することに成功しました。さらに、NNP-MDシミュレーションから得られた水素原子の自己拡散係数は、以前の第一原理MD(ab initio MD, AIMD)およびMLIP-MD計算結果と比較して、広範囲の含水率にわたって、実験値と良好な一致を示しました。
方法
本節では、ナフィオン系向けNNPモデルを構築する方法と、大規模ナフィオン系の長時間NNP-MDシミュレーションの実行条件について説明します。
NNPモデルの構築
NNPモデルの構築プロセスは図1の通りです。このプロセスの各段階について、以下に詳しく説明します。
初期構造の準備
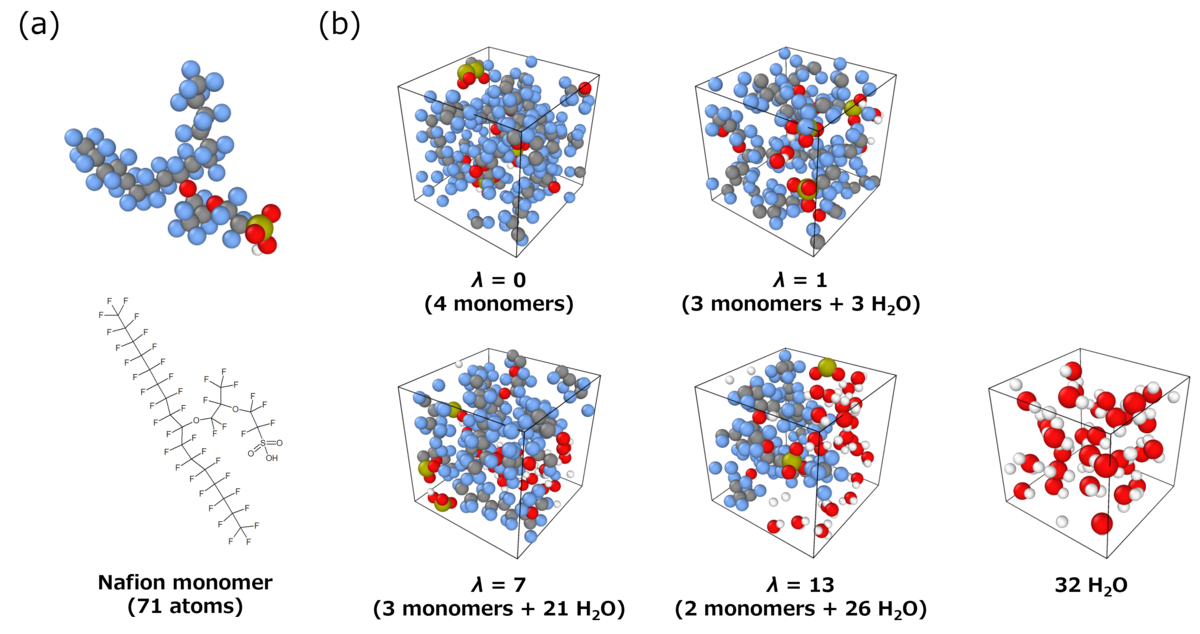
NNP訓練のための初期データ作成用の構造として、ナフィオンモノマーと水分子から構成される複数の系を使用しました。図2 (a) に示すように、ナフィオンモノマーは、炭素原子とフッ素原子で構成される疎水性のテフロン骨格と、スルホン酸基を持つパーフルオロ側鎖で構成され、合計71個の原子を含んでいます。このナフィオンモノマーと水分子から構成される初期構造として、図2 (b) に示す含水率λ = 0, 1, 7, 13の4構造を作成しました。ここで、λはスルホン酸基当りの水分子数として定義されます。これらの系は、それぞれ4個のナフィオンモノマー(λ = 0)、3個のモノマーと3個の水分子(λ = 1)、3個のモノマーと21個の水分子(λ = 7)、2個のモノマーと26個の水分子(λ = 13)で構成されています。さらに、32個の水分子からなる系も訓練構造に含めました。これらの初期構造の作成には、RadonPy [3] を用いました。
NNPの能動学習(AL)
図1にある通り、一つのALループは、NNP-MDによる構造サンプリング、モデルアンサンブルおよび構造特徴量に基づく2段階スクリーニング、DFTベースのラベリング、および増補されたデータセットを用いたNNPの再訓練という手順で構成されます。ALループの反復回数を増やすと、データセット内の構造多様性が向上します。本研究では、ラベル付き構造データを16,765個作成しました。なお最初のALループでは、AIMDシミュレーションから生成された初期データセットを使用して、初期のNNPを訓練しました。各段階の詳細を以下に説明します。
初期データセットの構築
最初のNNPの訓練データセットを生成するために、Quantum ESPRESSO [4] を用いて、図2 (b) に示した各系で、NVTアンサンブル下でAIMDシミュレーションを1ピコ秒実行しました。その際、50フェムト秒ごとにフレームを保存し、系ごとに200フレーム、合計1,000フレームを生成しました。各系のフレームを、8:2の比率で訓練データセットと検証データセットにランダムに分割しました。なお、各フレームには、全エネルギーと原子に作用する力のラベルを含めました。
NNPの訓練
NNPの訓練には、DeePMD-kit 2.2.4 [5] を使用しました。訓練ステップ数は、iteration 0–16では500,000、iteration 17では2,000,000としました。バッチサイズは1としました。学習率は、減衰ステップ数を5,000とし、1.0 × 10–3から1.0 × 10–8まで指数関数的に減衰する設定としました。損失関数は、エネルギー損失項と力損失項の加重和であり、各項の前因子は学習率に対して線形に変化する設定としました。また、独立して初期化された重みパラメータを持つ4つのDeep Potential (DP) モデルを訓練しました。これらの4つのDPモデルを使用して、以下のスクリーニングフェーズで原子に作用する力のモデル偏差 [6] を計算しました。
サンプリング
データセットに追加する構造をサンプリングするために、前のALループで生成されたDPモデルを用いて、図2(b)に示した各系でDPMDシミュレーションを実行しました。 初期のALループでは、NVTおよびNPTアンサンブル下でDPMDサンプリングを実行し、平衡状態近傍における構造を収集しました。その後、セル体積を徐々に圧縮する非平衡DPMD(non-equilibrium DPMD, NE-DPMD)サンプリングを実行しました。NE-DPMDサンプリングは、データセットに非平衡構造を導入することで、DPMDシミュレーションの安定性を向上させることを目的としています。圧縮シミュレーションは、原子間距離が短い構造を効率的にサンプリングすることを目的としています。 フレームは、各DPMDシミュレーションから異なる間隔で抽出しました。 DPMDシミュレーションの実行にはLAMMPS [7] を使用しました。
スクリーニング
サンプリングフェーズで取得された候補構造に対して、2段階のスクリーニングを適用しました。1段階目のスクリーニングでは、原子に作用する力の予測値のモデル偏差に基づいてスクリーニングを行いました。具体的には、4つのDPモデルを使用して、各構造について原子に作用する力を予測し、予測値の最大標準偏差が特定の範囲内に収まる構造のみを選択しました。これにより、現在のデータセットですでに十分に表現されている構造と、非常に不確実性が高く、非物理的である可能性が高い構造を除外できるため、NNPの精度を効率的かつ段階的に向上させることができます。
1段階目のスクリーニングを通過した構造に対して、構造特徴量に基づく2段階目のスクリーニングを適用しました。1段階目のスクリーニングを通過した構造は、多いときは数万個にも及びます。これらの構造群は、構造特徴量が類似したものを多く含んでおり、冗長な構造データです。2段階目のスクリーニングの目的は、構造特徴量空間内で近い距離にある構造を除外し、多様な構造特徴量を持つ候補構造を均一に選択することです。 ここでは、DPモデルの中間特徴量空間内の点間距離に基づいてスクリーニングを実行しました。本研究では、最大1,000個の構造が2段階目のスクリーニングを通過する設定としました。
ラベリング
スクリーニングフェーズで選択された最大1,000個の構造に対してDFT計算を実行し、全エネルギーと原子に作用する力のラベルを取得しました。DFT計算条件は、初期データセット生成時の条件と同じです。各系について、新しくラベル付けされたフレームを、8:2の比率で訓練データセットと検証データセットにランダムに分割しました。
以上のALを実行することで、訓練されたモデルの検証データセットにおけるエネルギーの二乗平均平方根誤差(Root Mean Squared Error, RMSE)は最大で2 meV/atom程度となりました。また、力のRMSEに関しても、ナフィオン系で最大65 meV/Å程度、水系で50 meV/Å未満となりました。エネルギーや力の予測精度という観点では、十分に高精度なDPモデルを構築できたといえます。
大規模NNP-MDシミュレーション
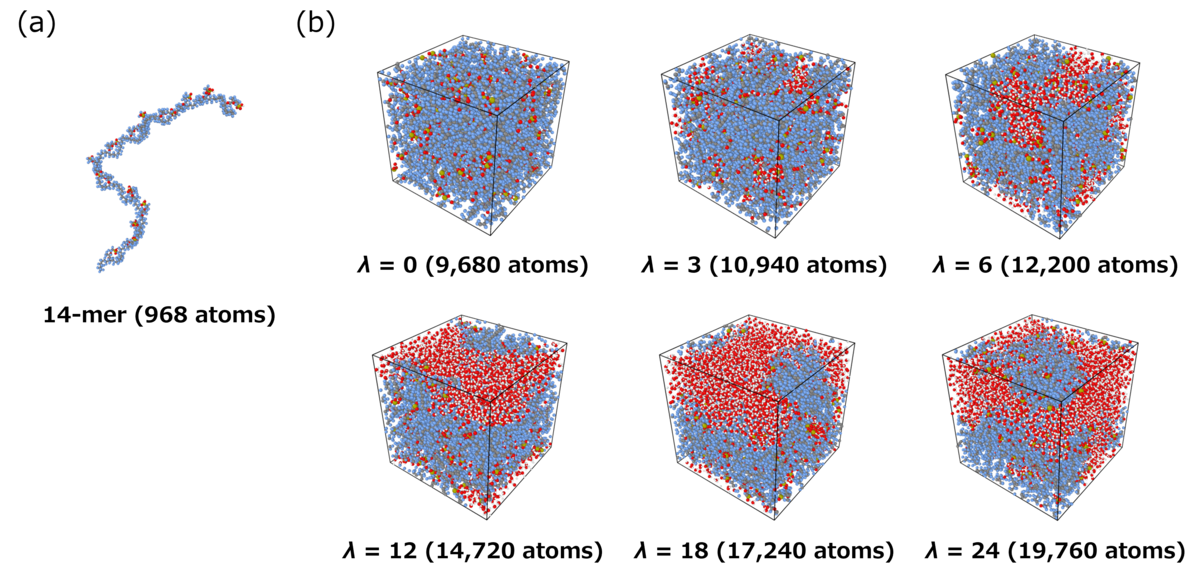
訓練済みのDPモデルを使用して、大規模ナフィオン系の長時間MDシミュレーションを実行しました。重合度14のナフィオンオリゴマー(図3 (a))を計算系に10個配置し、水分子を挿入することで、λ = 0、3、6、12、18、24のナフィオン系を作成しました(図3 (b))。各系の原子数は、それぞれ9,680、10,940、122,00、14,720、17,240、19,760です。なお、物理化学的特性の構造依存性を評価するために、各λで3つの独立した構造を作成しました。これらの初期構造の作成には、RadonPy [3] を用いました。
作成した初期構造を用いて、温度298 K、圧力1 barのNPTアンサンブル下でDPMDシミュレーションを実行しました。時間刻みは0.5フェムト秒で、シミュレーション時間は最大で31ナノ秒としました。各タイムステップで原子に作用する力のモデル偏差を評価するために、4つのDPを使用して原子に作用する力を予測し、予測値の標準偏差を計算しました。
結果と考察
本節では、図1に示すGeNNIP4MDのAL iteration x(x = 10, 13, 16, 17)で構築されたDPモデルを用いたMDシミュレーションの安定性について説明します。後述するように、GeNNIP4MDで構築されたDPモデルを用いることで、図3 (b) に示したすべてのナフィオン系で、30ナノ秒を超える長時間のMDシミュレーションが可能になることが分かりました。続いて、iteration 17で構築されたDPモデルを用いたMDシミュレーションにより得られた密度と水素原子の自己拡散係数を示します。なお、以降では、iteration x で構築されたDPモデルをDP-iter-x と書くこととします。
DPMDシミュレーションの安定性
図4は、DP-iter-10、-13、-16、および-17モデルのそれぞれについて、λ = 3のナフィオン系のMDシミュレーションの安定性をまとめたものです。DP-iter-10モデルを使用した場合、0.41ナノ秒付近で密度とポテンシャルエネルギーが急激に減少し、MDシミュレーションが崩壊しています。また、原子に作用する力の最大モデル偏差も0.41ナノ秒付近で急激に増加しています。このように、見かけ上のシミュレーション崩壊は0.41ナノ秒付近で起こっているものの、力の最大モデル偏差はそれよりもはるかに早い0.134ナノ秒付近で1 eV/Åを超えており、MDシミュレーションが初期の段階で不安定になっていることが分かります。したがって、長時間DPMDシミュレーションの安定性を確保するためには、力の最大モデル偏差の急激な増加を抑制することが不可欠です。
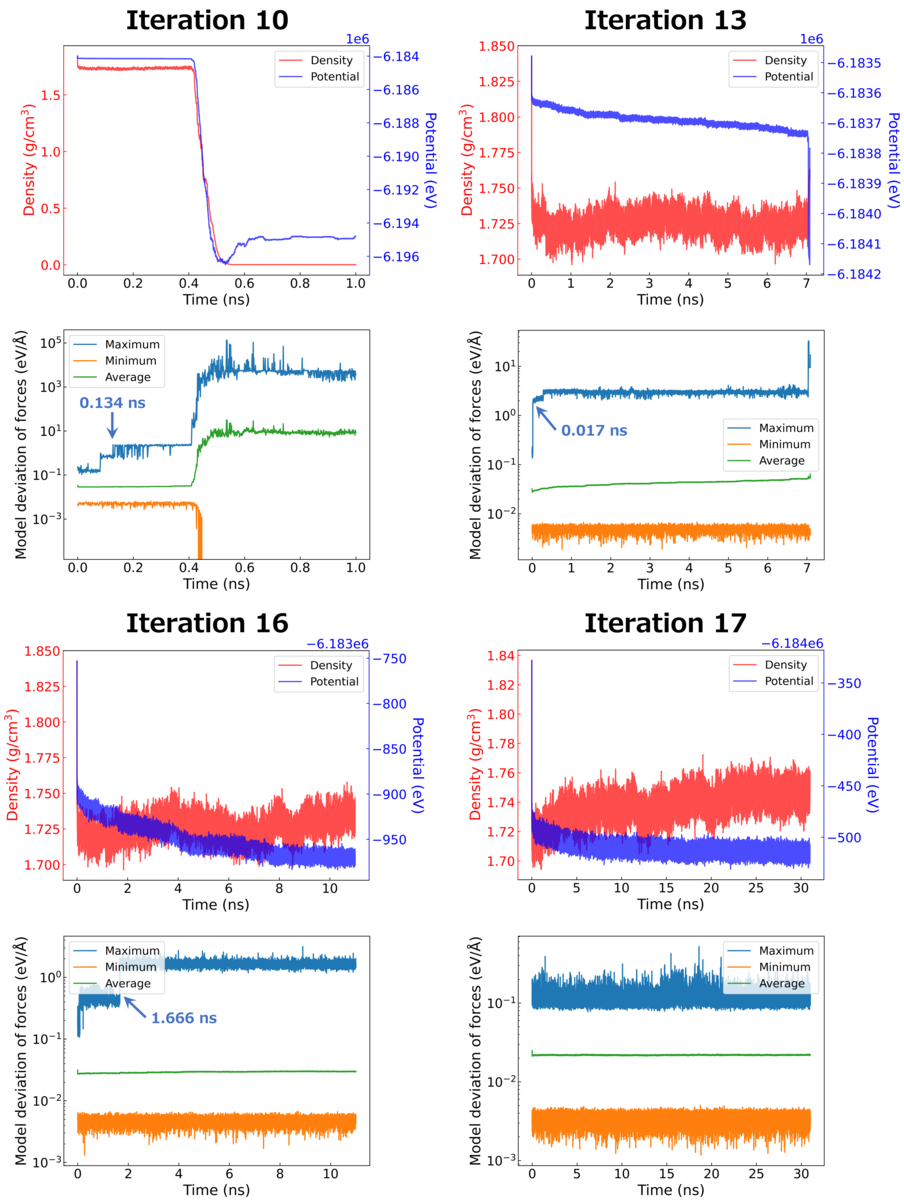
図4に示すように、DP-iter-13モデルを使用したMDシミュレーションは、密度とポテンシャルエネルギーの観点では、7ナノ秒付近まで安定しています。その後、ポテンシャルエネルギーの急激な減少および力の最大モデル偏差の急激な増加が起こり、シミュレーションが崩壊しています。ただし、力の最大モデル偏差はそれよりもはるかに早い0.017ナノ秒付近で1 eV/Åを超えており、ごく初期の段階でMDシミュレーションが不安定になっていることが分かります。
DP-iter-16モデルを使用したMDシミュレーションは、密度とポテンシャルエネルギーの観点では、10ナノ秒以上安定しています。力の最大モデル偏差は、1.666ナノ秒程度で1 eV/Åを超えていますが、DP-iter-10および-13モデルを使用した場合と比べて、MDシミュレーションの安定性は大幅に向上しています。力の最大モデル偏差の増加は、プロトンがナフィオンのエーテル酸素原子に誤って接近することに起因することが分かりました。このような原子環境を含む構造は非物理的であり、DFT計算がほとんど収束しないため、データセットに含めることができません。この問題に対処するために、水素原子とナフィオンのエーテル酸素原子の間に、内側カットオフ0.1 Å、外側カットオフ2 ÅのZiegler–Biersack–Littmark(ZBL)反発ポテンシャル [8] を導入しました。
DP-iter-17モデルを使用したMDシミュレーションは、31ナノ秒の長時間にわたって安定しています。さらに、力の最大モデル偏差は10–1 eV/Åのオーダーに留まっており、MDシミュレーションの安定性を示しています。他のすべてのλの場合でも、31ナノ秒の長時間にわたって、安定したMDシミュレーションを実行することに成功しました。これらの結果から、ナフィオン系の大規模・長時間MDシミュレーションを実行可能なDPモデルの構築に成功したといえます。
物理化学的特性
図5は、DP-iter-17モデルを用いたMDシミュレーションから得られた密度の計算値と実験値 [9] を比較したものです。密度の計算値は実験値よりも小さくなっているものの、幅広い含水率にわたって、密度の含水率依存性の傾向を再現できています。また、密度の計算値の標準偏差は十分小さく、大規模・長時間DPMDシミュレーションを実行することで、初期構造に対する依存性が小さく、信頼性の高い計算結果が得られたといえます。
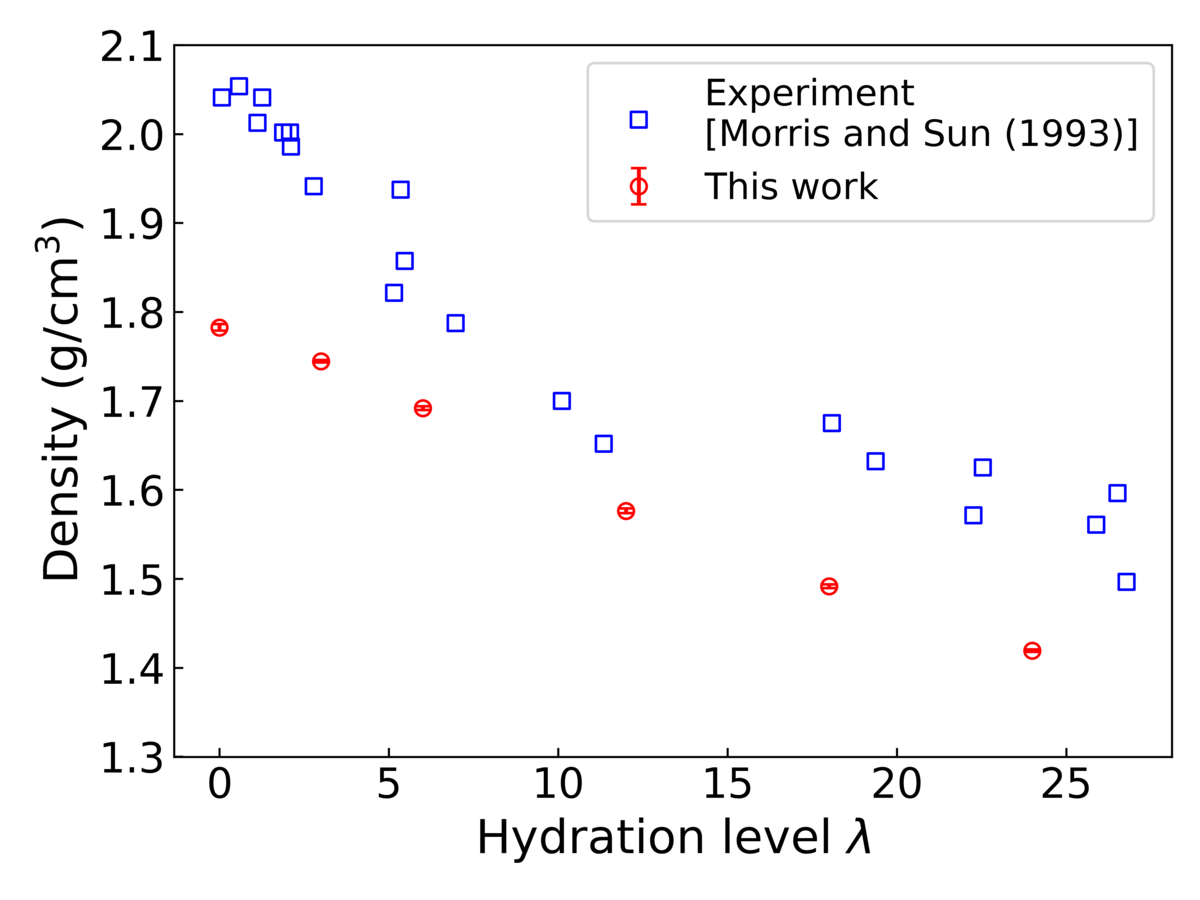
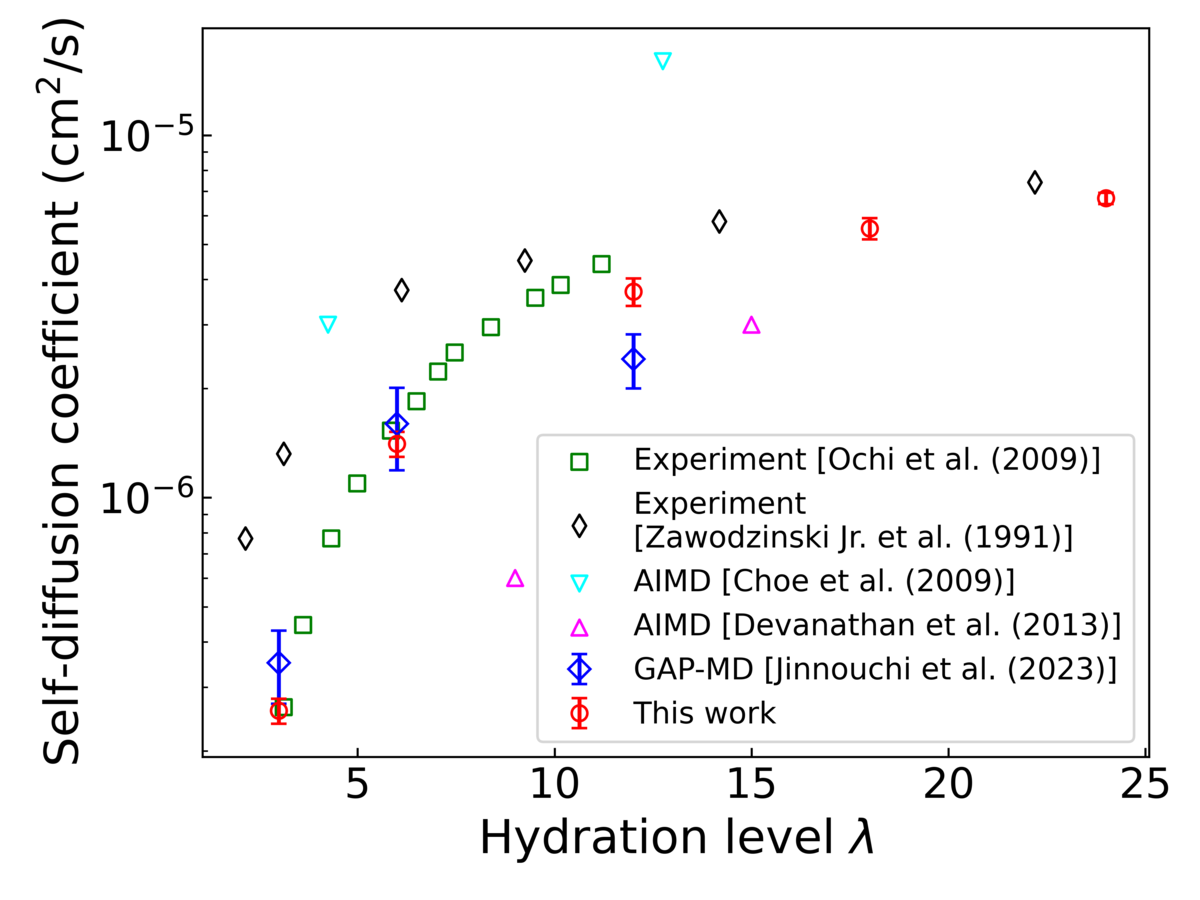
図6は、DPMDシミュレーションから得られた水素原子の自己拡散係数を先行研究の値と比較したものです。なお、自己拡散係数は、水素原子の平均二乗変位の傾きから算出しました。DPMD計算結果は、幅広い含水率にわたって、実験値 [10,11] とよい一致を示しています。一方で、以前のAIMDシミュレーション [12,13] は、計算系のサイズやシミュレーション時間の制約により、自己拡散係数を定量的には再現できていません。また、Gaussian Approximation Potential(GAP)を用いたMDシミュレーション [14] は、λ = 3および6では自己拡散係数をよく再現しているものの、λ = 12では自己拡散係数を過小評価しています。この結果は、GAP-MDの計算系のサイズ(約1,000原子)やシミュレーション時間(約200ピコ秒)は、不均質性の高い高含水率ナフィオン系における水素原子の輸送特性を再現するには不十分であることを示唆しています。一方で、私たちの大規模(>10,000原子)・長時間(>30ナノ秒)DPMDシミュレーションは、幅広い含水率にわたって、水素原子の輸送特性を精度よく再現できています。加えて、DPモデルの訓練に使用した構造(図2 (b))のλの最大値は13であるにも関わらず、それよりも大きなλの系(λ = 18, 24)においても、自己拡散係数をよく再現できています。このλに関する外挿性は、ALにより多様な局所原子環境をデータセットに含めることができたことに起因すると考えられます。
まとめ
本研究では、幅広い含水率のナフィオン膜の大規模・長時間MDシミュレーションを可能にする、ロバストなNNPモデルを構築しました。構築したDPモデルを用いることで、約10,000–20,000個の原子から成る大規模なナフィオン系の安定したMDシミュレーションを31ナノ秒の長期間にわたって実行することに成功しました。また、私たちのDPMDシミュレーションでは、以前のAIMDやGAP-MDシミュレーションと比較して、実験値により近い水素原子の自己拡散係数が得られました。今回ご紹介したGeNNIP4MDに搭載されているロバストなNNPを構築するためのAL技術は、幅広い材料に適用可能であり、大規模・長時間NNP-MDシミュレーションの安定実行を可能にするための有効な手法であるといえます。
参考文献
[1] Matsumura, N. et al. Generator of Neural Network Potential for Molecular Dynamics: Constructing Robust and Accurate Potentials with Active Learning for Nanosecond-Scale Simulations. J. Chem. Theory Comput. 2025, 21, 3832–3846.
[2] Yoshimoto, Y. et al. Large-Scale, Long-Time Atomistic Simulations of Proton Transport in Polymer Electrolyte Membranes Using a Neural Network Interatomic Potential. arXiv:2503.20412, 2025.
[3] Hayashi, Y. et al. RadonPy: Automated Physical Property Calculation Using All-Atom Classical Molecular Dynamics Simulations for Polymer Informatics. npj Comput. Mater. 2022, 8, 222.
[4] Giannozzi, P. et al. Quantum ESPRESSO toward the Exascale. J. Chem. Phys. 2020, 152, 154105.
[5] Zeng, J. et al. DeePMD-Kit v2: A Software Package for Deep Potential Models. J. Chem. Phys. 2023, 159, 054801.
[6] Zhang, Y. et al. DP-GEN: A Concurrent Learning Platform for the Generation of Reliable Deep Learning Based Potential Energy Models. Comput. Phys. Commun. 2020, 253, 107206.
[7] Thompson, A. P. et al. LAMMPS - a Flexible Simulation Tool for Particle-Based Materials Modeling at the Atomic, Meso, and Continuum Scales. Comput. Phys. Commun. 2022, 271, 108171.
[8] Ziegler, J. F. et al. The Stopping and Range of Ions in Matter; Pergamon, 1985; Vol. 1.
[9] Morris, D. R.; Sun, X. Water-Sorption and Transport Properties of Nafion 117 H. J. Appl. Polym. Sci. 1993, 50, 1445–1452.
[10] Zawodzinski, T. A. Jr. et al. Determination of Water Diffusion Coefficients in Perfluorosulfonate Ionomeric Membranes. J. Phys. Chem. 1991, 95, 6040–6044.
[11] Ochi, S. et al. Investigation of Proton Diffusion in Nafion®117 Membrane by Electrical Conductivity and NMR. Solid State Ion. 2009, 180, 580–584.
[12] Choe, Y.-K. et al. Nature of Proton Dynamics in a Polymer Electrolyte Membrane, Nafion: A First-Principles Molecular Dynamics Study. Phys. Chem. Chem. Phys. 2009, 11, 3892–3899.
[13] Devanathan, R. et al. Ab Initio Molecular Dynamics Simulation of Proton Hopping in a Model Polymer Membrane. J. Phys. Chem. B 2013, 117, 16522–16529.
[14] Jinnouchi, R. et al. Proton Transport in Perfluorinated Ionomer Simulated by Machine-Learned Interatomic Potential. J. Phys. Chem. Lett. 2023, 14, 3581–3588.
お問い合わせ
GeNNIP4MDにご興味をお持ちの方は、以下の連絡先までお気軽にお問い合わせください。
- 連絡先:fj-mi-tech-contact@dl.jp.fujitsu.com
- お問い合わせ内容:資料請求、技術紹介、PoC検証(技術の試用、自社材料への適用を希望される方)など、様々なご要望に対応いたします。