
はじめに
こんにちは、富士通研究所 コンピューティング研究所の西口和孝です。私たちはマテリアルズ・インフォマティクス(MI)の技術開発に取り組んでいます。
MIとは、AIをはじめとする情報科学の技術を活用し、材料開発の迅速化や新材料探索の効率化を行う技術手法です。 私たちが開発した分子動力学(Molecular Dynamics: MD)シミュレーション向けニューラルネットワークポテンシャル(Neural Network Potential: NNP)の自動生成ツールGeNNIP4MD (Generator of Neural Network Interatomic Potential for Molecular Dynamics)[1]は、量子力学に基づく第一原理計算のデータを学習し、高精度かつ低コストなシミュレーションが可能な機械学習ポテンシャルを生成するツールです(図1)。
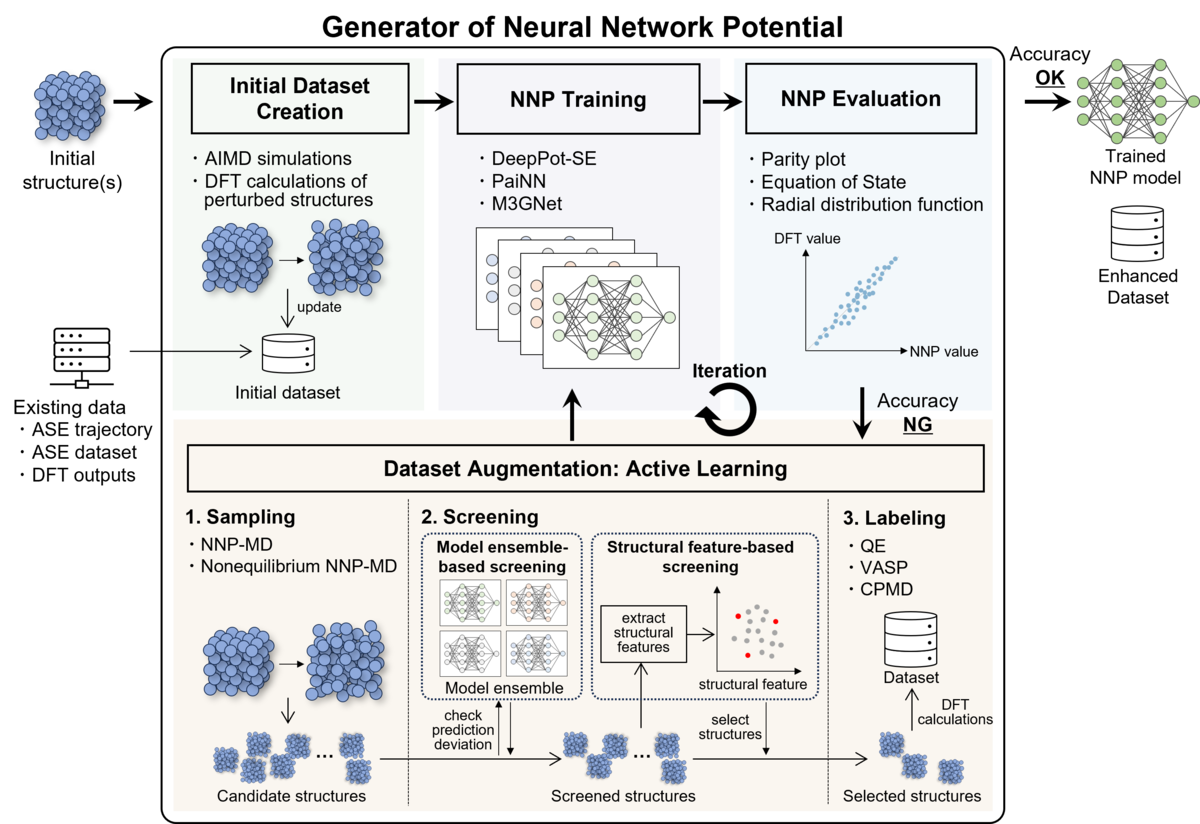
近年、NNPのような機械学習ポテンシャルは、通常の分子系や固体系だけでなくより複雑なアモルファス系や合金系、界面系にも応用が始まっています。 本記事では、GeNNIP4MD によって生成されたNNPを活用し、神戸大学様が実施されたSrTiO₃(チタン酸ストロンチウム)の酸素空孔の解析事例[2]を紹介します。
・GeNNIP4MDのSrTiO₃への適用事例
arxiv.org
・GeNNIP4MDの詳細
SrTiO₃の酸素空孔が材料特性に与える影響
本研究では、ペロブスカイト酸化物であるSrTiO₃(チタン酸ストロンチウム)に焦点を当てています。 SrTiO₃は、その高い誘電率、広いバンドギャップ、優れた熱安定性などから、コンデンサ、メモリ、触媒、太陽電池などの幅広い電子デバイス材料として活発に研究されています。
私たちの身の回りの固体材料は、理想的には規則的な原子配列を持つ「完全結晶」ですが、現実の材料合成プロセスでは、あるべき所に原子が存在しない原子空孔や不純物の混入といった「点欠陥」が必ず生じます。 これらの点欠陥は、特に半導体材料において、その電気伝導型(n型/p型)を決定するなど、材料の特性に極めて大きな影響を与えます。
SrTiO₃の場合、特に結晶構造中に存在する酸素空孔(Vo)が、その電気伝導特性に大きな影響を与えることが知られています。 酸素空孔は、物理化学的にSrTiO₃をn型半導体化させ、そのキャリア濃度や移動度に大きな影響を与えます。 そのため、酸素空孔の生成、拡散、および他の欠陥や不純物との相互作用を原子レベルで理解することは、SrTiO₃の物性を精密に制御し、高性能デバイスを開発するために不可欠な課題となっています。
従来のMDシミュレーションによる酸素空孔の解析の限界
酸素空孔といった点欠陥のダイナミクスを解明するための強力なツールが、MDシミュレーションです。MDシミュレーションは、原子間の相互作用力を計算し、ニュートンの運動方程式に従って原子の動きを追跡することで、時間とともに変化する系の挙動を予測します。
しかし、従来のMDシミュレーションには大きな課題がありました。
・第一原理分子動力学(ab initio MD: AIMD):量子力学に基づいた密度汎関数理論(DFT)を用いて原子間の力を計算するため、非常に高い精度で物質の電子状態や原子間相互作用を記述できます。その結果、実験結果ともよく一致する信頼性の高いシミュレーションが可能です。しかし、DFT計算は膨大な計算資源を必要とするため、シミュレーションできる原子数や時間スケールが極めて限定的です。そのため、材料中で実際に起こる欠陥の拡散や相転移といった現象を捉えることは困難です。
・古典分子動力学(古典MD):原子間の相互作用を経験的な関数(古典ポテンシャル)で記述するため、AIMDに比べてはるかに高速な計算が可能です。しかし、その精度は用いる古典ポテンシャルの質に大きく依存します。特定の物質や条件下でしか機能しない場合が多く、化学結合の変化や電子状態の複雑な変化を伴う現象、例えば欠陥の生成や特定の原子環境での相互作用を正確に記述することは困難でした。
このように、従来のMDシミュレーション手法には「精度は高いが計算が遅いAIMD」と「計算は速いが精度に限界がある古典MD」というトレードオフが存在しており、原子レベルでの深い理解に基づく材料設計の大きな障壁となっています。
このトレードオフが、材料科学におけるMDシミュレーションの適用範囲を制限していました。
ニューラルネットワークポテンシャルによるMDシミュレーション:精度と計算速度の両立
この長年の課題に、近年、機械学習(Machine Learning: ML)の進展が新たな光を当てています。 特に注目されているのが、ニューラルネットワークポテンシャル(NNP)に代表される機械学習原子間ポテンシャル(ML Interatomic Potential: ML-IAP)です。 NNPは、少数の高精度なDFTの計算結果を「教師データ」として学習することで、原子配置とエネルギー・力の関係を記述するポテンシャルを構築します。
NNPの最大の利点は、一度学習が完了すれば、その後のシミュレーションではニューラルネットワークの推論だけで原子間の力を予測できる点にあります。 この推論はDFTに比べて格段に高速でありながら、学習データの範囲内であればDFTに匹敵する高精度を維持できます。 これにより、NNPによるMDシミュレーション(NNP-MD)は「AIMDの精度」と「古典MDの計算速度」という、これまで両立が困難だった要素を高いレベルで融合することが可能になります。 NNP-MDは、大規模な系や長時間のシミュレーションにおいて、これまで不可能だった原子レベルでの現象解明を可能にする、まさにゲームチェンジャーとなりつつあります。
NNP-MDによるSrTiO₃酸素空孔エネルギーの解析
本研究は、このSrTiO₃の酸素空孔に関する長年の課題に対し、NNP-MDシミュレーションを適用することで、その酸素空孔エネルギーを高精度かつ効率的に予測する新たなアプローチを提示しました。 具体的には、以下のステップで検証を行いました。
1.高精度NNPモデルの構築:
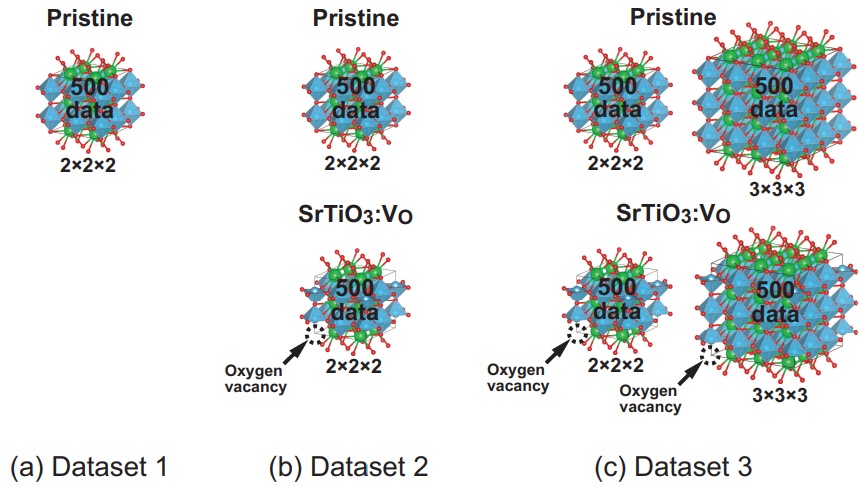
本研究では、純粋なSrTiO₃結晶と、単一の酸素空孔を含むSrTiO₃(SrTiO₃:V₀)の様々な超格子モデル(2x2x2、3x3x3)について、DFT計算によるラベリングにより、3種類の訓練データセットを生成しました(図2)。ラベルには、原子のエネルギーと原子にかかる力を含めました。
・データセット1:SrTiO₃結晶(2x2x2, 40原子) 500データ
・データセット2:SrTiO₃結晶(2x2x2) 500データ, 酸素空孔含むSrTiO₃(2x2x2, 39原子) 500データ
・データセット3:SrTiO₃結晶(2x2x2) 500データ, 酸素空孔含むSrTiO₃(2x2x2, 39原子) 500データ
SrTiO₃結晶(3x3x3, 135原子) 500データ, 酸素空孔含むSrTiO₃(3x3x3, 134原子) 500データ
NNPモデルの構築には、私たちの開発したNNP生成ツールであるGeNNIP4MD[1]を活用しました。 GeNNIP4MDは、アクティブラーニング(AL)と呼ばれる訓練手法を組み込んでいます。 GeNNIP4MDが搭載するALは、NNPモデルの予測精度が低い材料構造データを自動的に特定し、その構造データについて追加のDFT計算を行って学習データに加えるという反復プロセスです(図1)。 これにより、限られたDFT計算回数で、NNPモデルが広範囲の原子配置を正確に記述できるよう効率的に学習を進めることができます。 本研究では、このALプロセスを複数回反復することで、NNPモデルを構築しました。
2.構築したNNPモデルの検証:
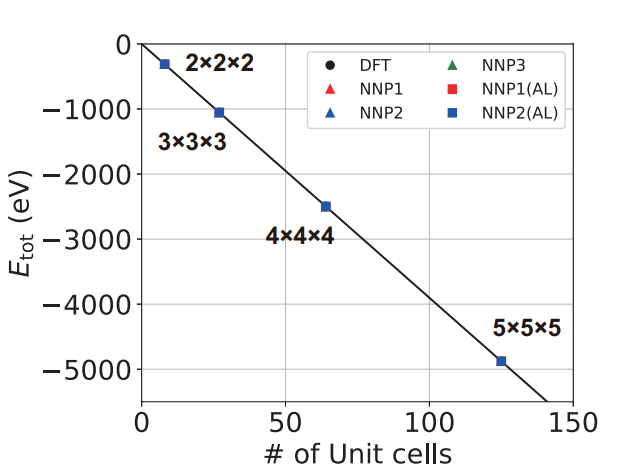
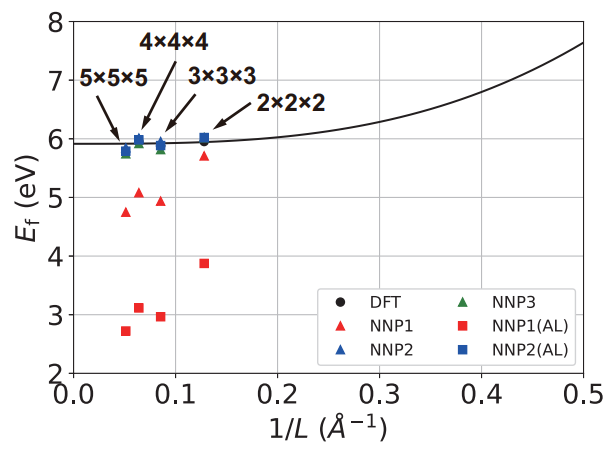
構築したNNPモデルの精度と信頼性を確認するため、NNP-MDシミュレーションによる予測結果と、DFT計算結果を比較しました(図3, 図4)。
・全エネルギーの予測: 様々なサイズの超格子モデル(2x2x2、3x3x3、4x4x4、5x5x5)におけるSrTiO₃の全エネルギーについて、NNPモデルの予測値はDFT計算値と非常に高い精度で一致しました。これは、構築したNNPモデルがSrTiO₃の基本的な原子間ポテンシャルを正確に表現できることを示しています。
・酸素空孔の形成エネルギーの予測: 材料の欠陥濃度を決定する上で重要な指標である酸素空孔の形成エネルギーについて、NNPモデルの予測精度を検証しました。 酸素空孔を含む訓練データ(データセット2, データセット3)を学習させたNNPモデル(NNP2, NNP3)は、DFT計算から得られた形成エネルギーを正確に再現できることが示されました。 特に、酸素空孔を含まない訓練データ(データセット1)を学習させたNNPモデル(NNP1)では形成エネルギーを予測できなかったことから、欠陥を含む系を正確に記述するためには、学習データにその欠陥の情報を適切に含めることの重要性が改めて確認されました。
3.大規模系へのNNP-MDの適用:
DFT計算では計算コストが高すぎて計算が困難であった、より大規模な超格子モデル(4x4x4, 5x5x5)における酸素空孔の形成エネルギーについても、NNP-MDシミュレーションを用いて予測を行いました。
図4に示すように、構築したNNPモデルは、これらの大規模系における形成エネルギーを、DFTの外挿値(有限サイズ効果を考慮して無限大の系に外挿した値)と極めて高い精度で一致させることができました。
この結果は、NNPが単に学習データ内の系を再現するだけでなく、DFTでは到達できないような大規模な系に対しても、その物理量を信頼できる精度で予測できるというポテンシャルを示しています。
これは、材料科学におけるMDシミュレーションの適用範囲を飛躍的に拡大できることを示唆しています。
まとめ
本研究の成果は、ニューラルネットワークポテンシャルに基づく分子動力学シミュレーションが、SrTiO₃のような複雑な結晶構造を持つ機能性材料において、全エネルギーや点欠陥の形成エネルギーといった重要な物理量を、高精度かつ大規模に予測できることを明確に実証しました。特に、DFT計算では困難であった大規模な系における欠陥の挙動を、信頼できる精度でシミュレーションできるようになった点は、材料科学におけるMDシミュレーションの適用範囲を飛躍的に拡大できることを示唆しています。
本研究は、SrTiO₃の酸素空孔の理解を深めるだけでなく、様々な機能性材料における点欠陥の挙動解明、さらには欠陥エンジニアリングによる新材料の設計・開発に大きく貢献するものです。例えば、NNP-MDを用いることで、以下のような研究が可能になります。
・欠陥の拡散経路と速度の解明: 長時間スケールのシミュレーションにより、欠陥が材料中をどのように移動するのか、その活性化エネルギーはどの程度かなどを詳細に解析できます。
・複雑な欠陥複合体の安定性評価: 複数の欠陥が集合した複合体の安定性や、それが材料物性に与える影響を評価できます。
・非平衡状態や相転移のダイナミクス: 熱処理や外部刺激によって引き起こされる材料の構造変化や相転移の過程を、原子レベルで追跡できます。
・新材料のスクリーニングと設計: 特定の機能を持つ材料を設計する際に、候補となる組成や構造における欠陥の挙動を事前に予測し、効率的な材料探索に貢献します。
本研究は、機械学習と物理シミュレーションの融合が、材料科学の未踏領域を切り拓く強力なアプローチであることを示しています。NNP-MDは、原子・分子レベルでの深い理解に基づいた、より効率的で精密な材料設計を可能にし、未来の技術革新を加速させるための強力な基盤となるでしょう。
お問い合わせ
GeNNIP4MDにご興味をお持ちの方は、以下の連絡先までお気軽にお問い合わせください。
- 連絡先:fj-mi-tech-contact@dl.jp.fujitsu.com
- お問い合わせ内容:資料請求、技術紹介、PoC検証(技術の試用、自社材料への適用を希望される方)など、様々なご要望に対応いたします。
参考文献
[1] N. Matsumura, et al., Generator of Neural Network Potential for Molecular Dynamics: Constructing Robust and Accurate Potentials with Active Learning for Nanosecond-Scale Simulations. J. Chem. Theory Comput. 2025, 21, 3832–3846.
[2] K. Nishiguchi, et al., Molecular Dynamics Simulations of SrTiO₃ with Oxygen Vacancies using Neural Network Potentials. arXiv:2506.09372, 2025.