
はじめに
こんにちは、富士通コンピューティング研究所 Materials Informatics Projectの山﨑です。私たちのチームでは、その名のとおりMaterial Informatics(MI)の研究開発を行い、材料技術に関するお客様の課題を解決することを目的して活動しております。 今回のMaterials Informatics特集 #4では、安定したMD(Molecular Dynamics: 分子動力学)シミュレーションが可能なMLIP((Machine Learning Interatomic Potential: 機械学習力場)について、最新の論文の知見をもとに紹介したいと思います。 なお、前回のMaterials Informatics特集 #3では、私たちの開発した高分子電解質膜向けニューラルネットワークポテンシャルについて紹介しましたので、ご興味のある方は下記のリンクをご参照ください。 blog.fltech.dev
突然ですが、皆さんはこのような経験をしたことはないでしょうか?
最近MD向けにAIを用いた機械学習力場が提案されていて、第一原理計算と同等の精度があるらしい...
でも使ってみようと調べたら、たくさんモデルが出てきてどのモデルを使ったら良いのかわからない
この記事では、このような課題を解決してくれる論文「Forces are not Enough: Benchmark and Critical Evaluation for Machine Learning Force Fields with Molecular Simulations」[1]を紹介したいと思います。
この論文では、SchNet[2]やDeepMD[3]などのMLIPが提案され始めた比較的初期ごろのモデルに加えて、PaiNN[4]、GemNet[5]、Nequip[6]などのより高精度なモデルを含む様々なMLIPモデルに対して、どのモデルがMDシミュレーションに適しているかを様々な事例で評価しています。 現実的な系に対して、様々なMLIPを用いたMDシミュレーションを実行し、どのモデルでのシミュレーションが安定しているか、また物性値をどの程度再現できるかを調べています。 最後まで読んでいただければ、どのMLIPモデルがMDに適しているか、またMDに適したモデルを判断する際にどのような指標で評価すべきかが理解できるようになりますので、ぜひ最後まで読んでいただけるとうれしいです!
既存のMLIPの評価の限界
前述のとおり材料科学の分野ではMLIPモデルが急速に注目を集めています。これらのモデルは、従来の経験的な力場に比べて、より高精度にMDシミュレーションを実行できるとされています。特に、SchNet[2]やDeepMD[3]などの初期のモデルから、PaiNN[4]、GemNet[5]、NequIP[6]といったより高精度なモデルまで、多様なMLIPが提案されてきました。これらのモデルは、主にDFT(Density Functional Theory: 密度汎関数理論) などの第一原理計算によって得られたエネルギーや力の値を基準に、RMSE(Root Mean Squared Error: 二乗平均平方根誤差)やMAE(Mean Absolute Error: 平均絶対誤差) などの指標でベンチマークされています。例えば、PaiNNの論文[4]では、QM9やMD17などのデータセットで、小分子に対するエネルギーや力、その他の物性値などのMAEの評価を行っています。
しかし、これらのエネルギーや力や指標だけでは、実際のMDシミュレーションにおけるモデルの有効性を十分に評価することはできません。エネルギーや力の誤差が小さく精度が高いことは、確かにPES(Poterntial Energy Surface: ポテンシャルエネルギー曲面)が多くの点で滑らかであることを示していますが、実際のMDシミュレーションで有効かどうかは別問題です。なぜなら、実際のMDシミュレーションはその名前のとおり動的であり、どのような状態に遷移するかを実行前に確かめることはできず、力の精度が高いのはあくまでもテスト用のデータに対してであって、実際の動的シミュレーション中の状態に対してではないからです。例えば、シミュレーション中に極端な力の予測が発生すると、システムが不安定になり、シミュレーションが瞬時に破綻する可能性があります。文献[7]では、MLIPを用いたプロピレングリコールのMDシミュレーションが行われていますこのシミュレーションでは、エネルギーのRMSEが小さなMLIPを用いても、実際にそのMLIPを用いたMDを実行すると0.1psの内にシミュレーションが崩壊してしまうことがわかっています。
このような背景から、MLIPモデルが実際にMDで使えるかを確かめるためには、単に力やエネルギーの予測精度を評価するだけでなく、シミュレーションの安定性や実際の物理的挙動を評価することが重要です。具体的には、短時間のMDシミュレーションを用いて、物性値などのシステムの特性を評価することが求められます。これにより、MLIPモデルが実際のMDシミュレーションにおいてどの程度有用であるかをより正確に判断することができます。
これがこの論文の主張するForces are not Enough(力の精度評価では十分ではない)ということであり、この論文ではこれを示すために、より現実的なシステムで幅広くシミュレーションを行い、テストデータに対する力の精度とシミュレーションの安定性、シミュレーションから計算される物性値を評価しています。
評価対象のモデル
本論文では以下の9つのMLIPモデルを用いて評価を行っています。異なるアーキテクチャ、計算コスト、表現力を持つモデルを網羅的に評価することで、MDシミュレーションにおけるMLIPの性能特性をより深く理解することを目的としています。
- DeepPot-SE[3]: シンプルな線形層と行列演算だけで設計されたモデル。表現力はそれほど高くないが高速に計算できる。
- SchNet[2]: メッセージパッシングを使用した初期のモデル。メッセージパッシングを使っているものの、回転不変な特徴量のみを使用しており軽量。
- DimeNet[8]: メッセージパッシングをする際に3原子の角度情報を用いるモデル。表現力は高いが、その分計算コストも高め。
- PaiNN[4]: 回転不変な特徴量と同変な特徴量を分けてメッセージパッシングを行うモデル。表現力と計算コストのバランスが良い。
- SphereNet[9]: 角度情報に加え、効率的に計算したねじれ角も考慮する。表現力と計算効率性を両立。
- ForceNet[10]: SchNetのメッセージパッシングをエッジ情報で拡張し、回転データの拡張を行って訓練されたモデル。計算量をあまり増加させずに表現力を向上させている。
- GemNet-T[5]: DimeNetを発展させてねじれ角も考慮したメッセージパッシングを可能にしたモデル。表現力は高いが計算速度が遅い。
- GemNet-dT[5]: GemNet-Tをベースに、力をエネルギーの微分ではなく、直接予測するようにしたモデル。エネルギーを微分しないので速度は速いが、エネルギーの保存性は担保されておらずシステムのエネルギーが収束しない可能性がある。
- NequIP[6]: ベクトルや高次のテンソルを含む特徴量を使用することで高い表現力を実現するモデル。少ない訓練データでも高い訓練精度を実現できる。
評価対象のシステム
本論文では、下のFigure 2に示されている4つのシステム((a) MD17, (b,c) 水, (d)アラニンジペプチド, (e)LiPS)を用いて評価が行われています。
 有機小分子を扱うMD17、基本的な系である水(H2O)、生体分子をカバーするアラニンジペプチド、無機系の電池材料のLiPSと様々なシステムを使用することで、幅広くモデルの性能を評価しています。今回の記事では、MD17とH2Oの結果を中心に紹介します。
アラニンジペプチドでは、ほぼ全てのモデルで力のMAEが大きく、安定したMDシミュレーションを実行することが難しい場合が多いためです。逆にLiPSでは、ほぼ全てのモデルで力のMAEが小さく、安定したMDシミュレーションを実行できたため、本記事ではMD17とH2Oの結果を中心に紹介します。
有機小分子を扱うMD17、基本的な系である水(H2O)、生体分子をカバーするアラニンジペプチド、無機系の電池材料のLiPSと様々なシステムを使用することで、幅広くモデルの性能を評価しています。今回の記事では、MD17とH2Oの結果を中心に紹介します。
アラニンジペプチドでは、ほぼ全てのモデルで力のMAEが大きく、安定したMDシミュレーションを実行することが難しい場合が多いためです。逆にLiPSでは、ほぼ全てのモデルで力のMAEが小さく、安定したMDシミュレーションを実行できたため、本記事ではMD17とH2Oの結果を中心に紹介します。
MD17
MD17データセットは有機小分子のデータセットで、アスピリンやエタノール等の8種類の分子で第一原理計算MDを実行した時のトラジェクトリーで構成されています。今回の評価では、アスピリン、エタノール、ナフタレン、サリチル酸の4種類の分子を使用し、それぞれのトラジェクトリーから訓練データを9,500個,、検証データを500個、テストデータを10,000個のランダムに抽出し使用します。MDシミュレーションは、ノセフーバーサーモスタットを使用して500Kで0.5fs間隔で300ps、テストデータからサンプルされた5つの初期構造で実行します。評価指標は、テストデータに対する力のMAEに加えて、原子間距離分布h(r)のMAEを評価します。また、シミュレーションの安定性(Stability)を、原子間距離のMAEの最大値が0.5Åを超えることなくシミュレーションが継続した時間を計測することで評価します。
H2O
H2Oのデータセットは、1nsのMDシミュレーションから10fs間隔で抽出された100,000個のデータで構成されます。訓練データと検証データの合計数は1,000/10,000/90,000の3パターンで、テストデータは10,000個で固定です。MDシミュレーションは、ノセフーバーサーモスタットを使用して300Kで1fs間隔で64分子のシステムでは500ps、150分子のシステムでは150ps、テストデータからサンプルされた5つの初期構造で実行します。評価指標はテストデータに対する力のMAEに加えて、各元素ペアのRDF(Radial Distribution Function: 動径分布関数)FのMAEと自己拡散係数のMAEを使用します。安定性(Stabilitiy)の定義がMD17の時とは少し変わり、RDFがのMAEの値が1.0を超えることなくシミュレーションが継続した時間となっています。
評価結果
MD17の評価結果
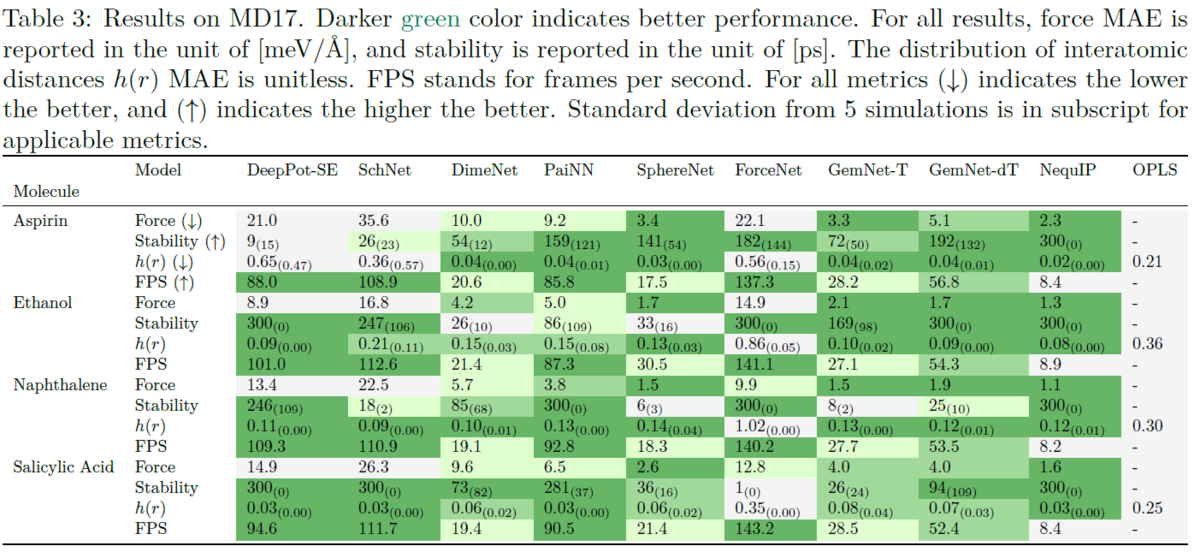 MD17の結果は上のTable 3にまとめられています。
Table 3は、MD17データセットにおける各モデルの力のMAE、安定性(Stability)、原子間距離分布h(r)のMAE、FPS(Frames per Second: フレームレート)を示しています。
NequIPについては、全ての分子において力とh(r)のMAEが小さく、さらにStabilityの結果から5つの初期構造によるシミュレーションがすべて崩壊することなく300ps実行できていることがわかります。一方でNequIPにも弱点があり、FPSが小さいことから計算速度が非常に遅いことがわかります。PaiNNはNequIPと比較すると力やh(r)のMAEは小さくなくStabilityの値も大きくはないです。しかしながら他のモデルと比較した場合には、特別に精度や安定性が低いわけではないです。FPSは80~90になっているため、速度の精度のバランスが良いと言えます。
MD17の結果は上のTable 3にまとめられています。
Table 3は、MD17データセットにおける各モデルの力のMAE、安定性(Stability)、原子間距離分布h(r)のMAE、FPS(Frames per Second: フレームレート)を示しています。
NequIPについては、全ての分子において力とh(r)のMAEが小さく、さらにStabilityの結果から5つの初期構造によるシミュレーションがすべて崩壊することなく300ps実行できていることがわかります。一方でNequIPにも弱点があり、FPSが小さいことから計算速度が非常に遅いことがわかります。PaiNNはNequIPと比較すると力やh(r)のMAEは小さくなくStabilityの値も大きくはないです。しかしながら他のモデルと比較した場合には、特別に精度や安定性が低いわけではないです。FPSは80~90になっているため、速度の精度のバランスが良いと言えます。
次に、興味深い結果として、DeepPot-SEがあげられます。DeepPot-SEはアスピリン以外の分子では、Stabilityの値が大きく、h(r)のMAEは小さいのでシミュレーションが安定して実行できていることがわかります。しかし、力のMAEはSchNetに次いで大きいです。 一方、SphereNetやGemNet、ForceNetは力のMAEの値が小さいのにも関わらず、Stabilityの値が小さくシミュレーションが安定して実行できていない場合が多いです。 つまり、MD17のテストデータに対して力のMAEが小さくなるように最適化したとしても、MDシミュレーションが安定的に実行できるかはわからないということです。 これらの結果は、MDシミュレーションにおけるMLIPの性能を評価する際には、テストデータに対する力のMAEだけでなく、シミュレーションの安定性や物性値の再現性も考慮する必要があることを示唆しています。
H2Oの評価結果
 次に、H2Oのシステムでの評価結果を見てみましょう。
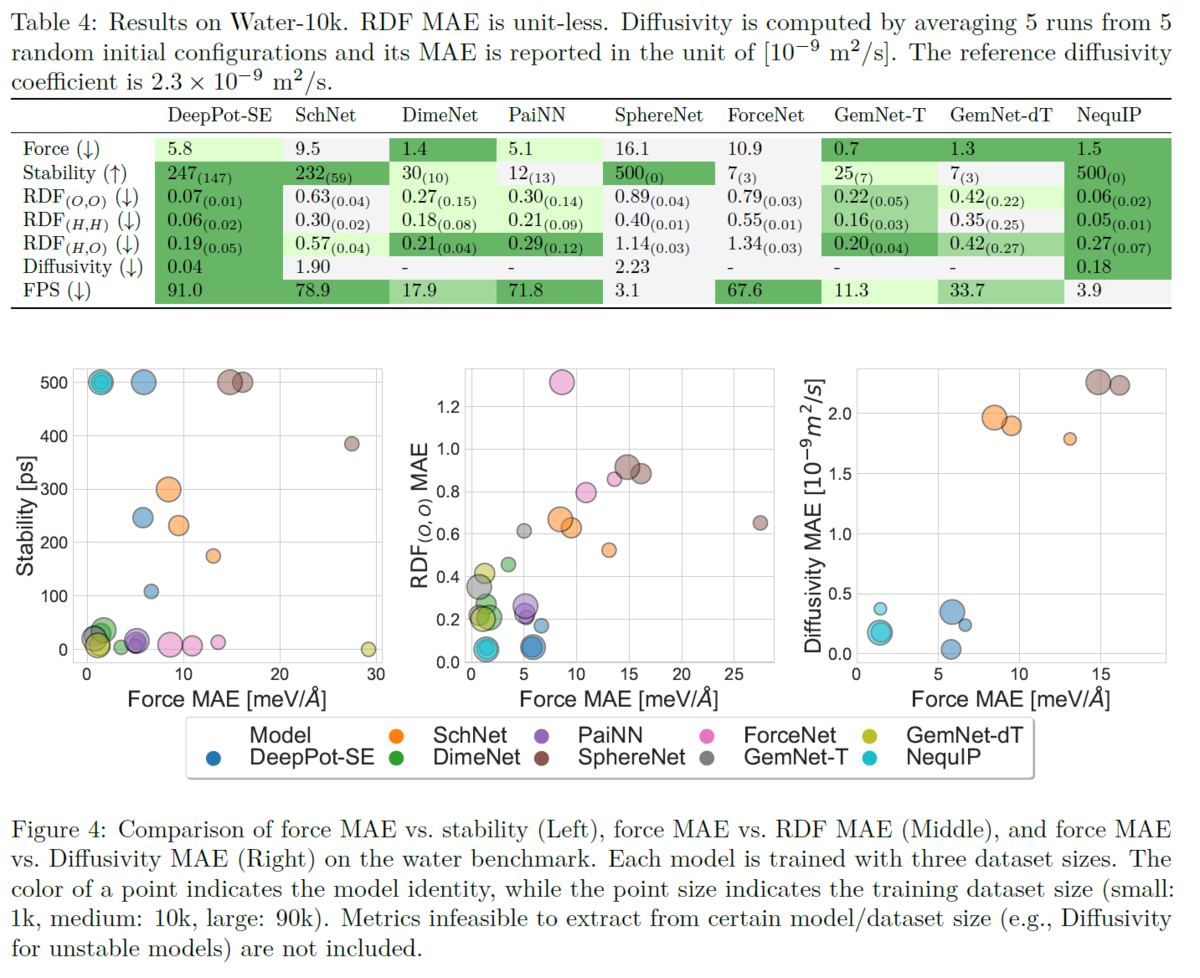
上のTable4は、H2Oデータセットを用いたMLIPの評価結果となります。
Table 4は、H2Oデータセットにおける各モデルの力のMAE、安定性(Stability)、RDF、自己拡散係数(Diffusivity)、FPS(Frames per Second: フレームレート)を示しています。
MD17の時と同様に、H2OデータセットでもNequIPが精度やStabilityが非常に高いですが、一方で計算速度が遅いことがわかります。
また、DeepPot-SEもMD17を用いた場合と似た結果となっており、力のMAEは大きいですが、小さなRDFのMAEと高いStabilityを実現しています。
DimeNetやGemNetは力のMAEが最も小さいにもかかわらず、Stabilityが低いために、すぐにシミュレーションが破綻してしまうことが分かります。
従って、H2Oのシステムにおいても力のMAEと、シミュレーションの安定性や物性値のMAEの関連が薄いことがわかります。
次に、H2Oのシステムでの評価結果を見てみましょう。
上のTable4は、H2Oデータセットを用いたMLIPの評価結果となります。
Table 4は、H2Oデータセットにおける各モデルの力のMAE、安定性(Stability)、RDF、自己拡散係数(Diffusivity)、FPS(Frames per Second: フレームレート)を示しています。
MD17の時と同様に、H2OデータセットでもNequIPが精度やStabilityが非常に高いですが、一方で計算速度が遅いことがわかります。
また、DeepPot-SEもMD17を用いた場合と似た結果となっており、力のMAEは大きいですが、小さなRDFのMAEと高いStabilityを実現しています。
DimeNetやGemNetは力のMAEが最も小さいにもかかわらず、Stabilityが低いために、すぐにシミュレーションが破綻してしまうことが分かります。
従って、H2Oのシステムにおいても力のMAEと、シミュレーションの安定性や物性値のMAEの関連が薄いことがわかります。
H2Oのシステムでは訓練データの数を変化させた評価も行っています。その結果が上に示すFigure 4です。 Figure 4は、左から順に、H2Oシステムにおける各モデルの力のMAEと、Stability, RDFのMAE, および水の自己拡散係数のMAEに対する訓練データ数の影響を示しています。円が大きいほど訓練データ数が多いことを示しています(小:1,000個, 中:10,000個, 大:90,000個)。 この図から、概ね全てのモデルで訓練データ数を増加すると力のMAEは小さくなることがわかります。しかし、Stabilitiyは必ずしも向上していません。わかりやすい例として、ピンクの円で表現されているForceNetは訓練データ増えることで、MAEが15meV/Åから8meV/Å程に改善されていますが、Stabilityは0付近からほとんど変化していません。このように、データ数の増加に伴ってシミュレーションのStabilityが向上しないモデルも存在する一方で、データ数の増加に伴ってStabilityが向上するDeepPot-SE(青の円)や、データ数が少ない段階ですでにStabilityが高いNequIP(水色の円)なども存在します。
最後に
この論文から、ベンチマーク用のデータセットで良い精度を達成しても、必ずしも安定したMDシミュレーションができるとは限らないことがわかりました。例えば、DimeNetやGemNetは、MD17およびH2Oのテストデータに対して高い力の予測精度を示しましたが、シミュレーションの安定性は低く、すぐに破綻してしまうことが本論文では示されています。評価結果を比べると、より精度を求めるならNequIPのような高次のテンソルを含む特徴量を持つモデルを使用するのが良いでしょう。ただし、計算コストが高いために、大規模な系や長時間のシミュレーションには不向きです。一方で速度と予測精度のバランスを考えるならDeepPot-SEのような軽量なモデルを使用するのが良いことがわかります。しかしながら、複雑な系や高精度な計算が必要な場合には精度が不足する可能性があります。
また、MD17およびH2Oの結果から、MLIPの性能評価においては、テストデータに対する力のMAEだけでなく、シミュレーションの安定性や物性値の再現性を総合的に評価する必要があることが改めて示されました。さらに、最後のH2Oの結果から、データセットを増やしても、シミュレーションの安定性が必ずしも向上するとは限らないことがわかりました。モデルのアーキテクチャや訓練方法によって、シミュレーションの安定性に対する訓練データ数の影響が異なることから、今後は、特定の系に対して高い精度と安定性を両立できるモデルを開発するために、モデル設計と訓練方法の両面から検討する必要があると考えられます。
私たちは、より安定なMDを実行するために訓練データの多様性が重要であると考えています。そして、安定なMDシミュレーションを実現するために、自動で多様なデータを生成し、MLIPモデルを訓練するツールとしてGeNNIP4MD[7]を開発しています。GeNNIP4MDは様々な条件下でMDシミュレーションを行い、そのトラジェクトリーから既存のデータに含まれない有望なデータを能動学習により見つけることで、データセットの多様性を向上させます。これにより安定性の高いMLIPモデルを構築することができ、大規模・長時間のMDシミュレーションを可能にします。GeNNIP4MDの詳細については、以下のFujitsu Tech blogのMaterials Informatics特集 #1や、参考文献 [1]をご参照ください。GeNNIP4MDにご興味がございましたら、下記のお問い合わせ先までお気軽にご連絡いただけると幸いです。
お問い合わせ
- 連絡先:fj-mi-tech-contact@dl.jp.fujitsu.com
- お問い合わせ内容:資料請求、技術紹介、PoC検証(技術の試用、自社材料への適用を希望されてる型)など、様々なご要望に対応いたします。
参考文献
[1] X. Fu et al. Forces are not Enough: Benchmark and Critical Evaluation for Machine Learning Force Fields with Molecular Simulations. Transactions on Machine Learning Research, 2023.
[2] K. Schütt et al. Schnet: A continuous-filter convolutional neural network for modeling quantum interactions. Advances in neural information processing systems, 30, 2017.
[3] L. Zhang et al. Deep potential molecular dynamics: a scalable model with the accuracy of quantum mechanics. Physical review letters, 120(14):143001, 2018a.
[4] K. Schütt et al. Equivariant message passing for the prediction of tensorial properties and molecular spectra. In International Conference on Machine Learning, pp. 9377–9388. PMLR, 2021.
[5] J. Gasteiger et al. Gemnet: Universal directional graph neural networks for molecules. Advances in Neural Information Processing Systems, 34:6790–6802, 2021.
[6] S. Batzner et al. E (3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials. Nature communications, 13(1):1–11, 2022.
[7] N. Matsumura et al. Generator of Neural Network Potential for Molecular Dynamics: Constructing Robust and Accurate Potentials with Active Learning for Nanosecond-scale Simulations. arXiv preprint arXiv:2411.17191, 2024. https://arxiv.org/abs/2411.17191.
[8] J. Gasteiger et al. Directional message passing for molecular graphs. In International Conference on Learning Representations, 2020.
[9] Y. Liu et al. Spherical message passing for 3d molecular graphs. In International Conference on Learning Representations, 2021.
[10] W. Hu et al. Forcenet: A graph neural network for large-scale quantum calculations. arXiv preprint arXiv:2103.01436, 2021.